
Д. Тяжелая комбинированная иммунная недостаточность, обусловленная дефицитом JAK-3
Группа 1. Комбинированные Т- и В-клеточные иммунодефициты
Клинические проявления: наследуются преимущественно по аутосомно-рецессив-ному типу (за исключением связанной с Х-хромосомой ТКИД).
Клинические симптомы проявляются в первые недели жизни в виде некор-ригируемой диареи, бактериальных и грибковых поражений кожи и слизистых оболочек, прогрессирующего поражения респираторного тракта, пневмоцистной пневмонии, вирусных инфекций, гипоплазии лимфоидной ткани. Пневмонию час-то вызывает Pneumocystis jiroveci, диарею – ротавирусы, Campilobacter, Giardia lamblia. Нередко развивается вирусный гепатит, в некоторых случаях – осложне- ние в виде БЦЖита после вакцинации БЦЖ в роддоме. Дети отстают в физичес-ком и психомоторном развитии. В ряде случаев при наличии ТКИД симптомы раз-виваются не сразу, в течение 2-6 мес жизни они могут расти и развиваться сравни- тельно нормально.
Основные критерии, позволяющие заподозрить комбинированный ПИД:
-наличие тяжелых, часто летальных инфекций, начиная с периода новорожденнос-ти; у больных развиваются затяжные формы диареи, пневмонии, сепсис;
-характерно инфицирование маловирулентными микроорганизмами, такими, как грибы рода Candida, пневмоцисты, ЦМВ;
-появление с рождения кожной сыпи в виде эритродермии, обусловленной мате-ринскими лимфоцитами, которые поступили к плоду во время беременности;
-отставание ребенка в росте и массе.
Лабораторные критерии– нарушение клеточного и гуморального имму-нитета с 6-7-месячного возраста: лимфопения (менее 4000 кл/мкл), снижение уров-ня IgG, IgM, IgA в сыворотке крови, отсутствие специфических АТ после вакци-нации, снижение или отсутствие Т- и/или В-лимфоцитов в зависимости от формы ИД.
Гистологическая картина – резкое снижение массы тимуса, отсутствие ти-моцитов, плохо дифференцированная кортикальная зона тимуса. Граница между кортикальной и медуллярной зонами смазана, а иногда и вовсе отсутствует, тель-ца Гассаля не развиты. Периферические лимфоидные органы гипоплазированы или отсутствуют. Лимфатические узлы, селезенка, миндалины, пейеровы бляшки зачастую находятся в стадии обратного развития, а лимфоциты в них выявить не удается. В лимфатических узлах отсутствуют герминативные центры.
Принципы лечения:
-трансплантация гаплоидентичного костного мозга или костного мозга, взятого от полностью совместимого по HLA донора, причем перед трансплантацией из до-норского костного мозга удаляют Т-лимфоциты, чтобы избежать реакции «транс-плантат против хозяина»
-заместительная терапия внутривенными иммуноглобулинами, интенсивное анти-бактериальное, противогрибковое и противовирусное лечение.
Прогноз — неблагоприятный. В большинстве случаев заболевание заканчива-ется летальным исходом в раннем возрасте ввиду развития генерализованных оп-портунистических инфекций.
А. Х-сцепленная тяжелая комбинированная иммунная недостаточность.
Гены, кодирующие ТКИД, картированы в положении Хq13 (60% случаев). В боль-шинстве случаев это точечные мутации, нарушающие цитокиновую регуляцию процессов развития и взаимодействия лимфоцитов. Происходят мутации генов, кодирующих общую γ- цепь рецепторов ИЛ-2, ИЛ-7, ИЛ-9, ИЛ-15, ИЛ-21. Проис-ходит нарушение взаимодействия ИЛ-7, секретируемого стромальными клетками КМ и тимуса, который служит ростовым фактором Т- и В-лимфоцитов, и его ре-цептора, нарушаются процессы дифференцировки и созревания Т- и В-лф.
При обследовании Т- и NK-клетки обнаружить не удается или их содержание мало, а содержание В-лф повышено. При этом наблюдается выраженная недоста-точность функций Т- и В-клеток. Подавление синтеза иммуноглобулинов сохраня-ется даже после восстановления нормального уровня Т-лф после пересадки КМ.
Диагностика:
-генетическое исследование ДНК на наличие мутаций в Хq13.1
-определение экспрессии общей γ -цепи на лф с помощью моноклональных анти- γ -антител
-пренатальная диагностика путем исследования ворсин хориона или клеток амнио-тической жидкости на наличие данной мутации либо исследование крови плода, начиная с 17-й недели развития, и выявление лимфопении за счет снижения коли-чества Т-клеток и их ответа на митоген.
Болеют только мальчики, клинические проявления возникают с 3-6 мес.
Б. Тяжелая комбинированная иммунная недостаточность с дефицитом адено-зиндезаминазы.
Возникает дефект гена фермента АДА, который расположен на длинном плече хромосомы 20, в локусе q12-13.1. В отсутствие АДА токсичные метаболиты пу-ринового обмена (дАТФ) и метилирования (S-аденозил-гомоцистеин) накаплива-ются внутри клетки и тормозят пролиферацию. В результате нарушается функция Т- и В- лф. Накопление дАТФ происходит и в эритроцитах. В плазме повышается концентрация аденозина и дезоксиаденозина.
Клиническая картина: ранняя манифестация – к 2-3-месячному возрасту появляются респираторные инфекции, диарея, отставание в развитии, множест-венные дефекты костей (хондродисплазия), затрагивающие костно-хрящевые сое-динения, апофизы подвздошных костей и тела позвонков (феномен «кость в кос-ти» на рентгене).
Поздняя манифестация –у детей от 2 до 15 лет развиваются повторные пнев-монии, вызванные S.pneumoniae, сепсис, заболевания аутоиммунной природы (СД, тиреоидит, гемолитическая анемия, идиопатическая тромбоцитопения), у взрос-лых рецидивирует герпетическая инфекция.
Диагностика:
-определение АДА в эритроцитах, лф, фибробластах
-обнаружение дАТФ и дезоксиаденозина в моче
-генетическое обследование на наличие мутации гена, кодирующего АДА
-пренатальная диагностика ворсин хориона, амниотических клеток крови плода на наличие мутаций гена, кодирующего АДА.
Лечение:
-трансплантация КМ от HLA-совместимого донора
-ферментозамещающая терапия 3 раза в неделю в виде подкожных инъекций пре-парата АДА крупного рогатого скота, конъюгированного с полиэтиленгликолем (препарат ADAGEN)
-генная терапия
В.Тяжелая комбинированная иммунная недостаточность с дефицитом пу- риннуклеозидфосфорилазы
Развивается в результате дефекта гена, находящегося на хромосоме 14 (локус q13.1). При отсутствии данного фермента токсичные метаболиты (дезоксигуано-зинтрифосфатаза (дГТФ)) накапливаются внутри клетки и нарушают пролифе-рацию лф. Т-лф особенно чувствительны к накоплению дГТФ, чем В-лф. В сыво-ротке крови накапливаются инозин, гуанозин, которые выделяются с мочой.
Лабораторные проявления
Клинические проявления: рецидивирующие вирусно-бактериальные инфек-ции, поражающие органы дыхания, ЛОР-органы, мочевыделительную систему, наблюдаются неврологические нарушения (диплегия, тетрапарез, атаксия, тремор, задержка умственного развития), возможно появление лимфомы и аутоиммунных расстройств.
Диагностика:
-исследование активности фермента в лизате эритроцитов (спектрофотометрия, радиохимический анализ)
-определение содержания мочевой кислоты в сыворотке крови (снижено)
-определение содержания инозина и гуанозина в сыворотке крови
-определение содержания инозина, гуанозина, дезоксиинозина и дезоксигуанозина в моче
-пренатальная диагностика путем исследования активности фермента и наличия мутации его гена в ворсинах хориона.
Г. Синдром Оменна
Причина – мутации в генах RAG1и RAG2, которые находятся на хромосоме 11р13 и участвуют в процессах соматической перестройки генов TCR и BCR. При мута-ции не формируется разнообразие TCR и BCR, а также АТ, нарушается экспрессия антигенных рецепторов вследствие торможения рекомбинации генов.
Клинические проявления: генерализованная эритродермия, десквамация кожных покровов, диарея, гепатоспленомегалия, лимфаденопатия, лихорадка.
Лабораторные проявления: очень низкое содержание CD19+ В-клеток, IgA, IgM, IgG значительно снижены, образование антител in vivo нарушено; лейкоци-тоз, гиперэозинофилия, повышено содержание IgE.
В лимфоузлах отсутствуют фолликулы с центрами размножения. Тимус гипо-плазирован.
Лечение – трансплантация КМ.
Прогнознеблагоприятный.
Полные мутации этих генов приводят к тотальному блоку развития Т- и В-клеток и вызывают ТКИД с отсутствием зрелых форм Т- и В-лф (пре-Т- и пре-В-клетки не выживают при дифференцировке, если не получают сигнал от пре-BCR и пре-TCR соответственно).
Д. Тяжелая комбинированная иммунная недостаточность, обусловленная дефицитом JAK-3
JAK3-киназа – единственная сигнальная молекула, взаимодействующая с γ-цепью, дефект гена этой киназы служит причиной развития аутосомно-рецессивного ва-рианта ТКИД.
В крови повышено количество В-лф, количество Т-лф и NK-клеток резко снижено
(иммунные сдвиги обусловлены наличием аномального рецептора для цитокинов на поверхности В-клеток).
Лечение — трансплантация КМ.
stydopedia.ru
Ингибиторы киназы JAK для лечения острого лимфобластного лейкоза — PubMed на русском
JAK kinase inhibitors for the treatment of acute lymphoblastic leukemiaИсточник: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4545857/
Недавние исследования острой лимфобластной лейкемии выявили активирующие мутации в компонентах рецепторного комплекса интерлейкина-7 (IL7R, JAK1 и JAK3). Будет интересно исследовать как ингибиторы киназы JAK1, так и JAK3 в качестве целевых агентов для этих лейкозов.
Цитокиновая сигнализация, организованная различными лигандами и более чем 30 различными рецепторами, играет критическую роль во время гемопоэза. Цитокины, которые связываются с рецепторами, содержащими общую гамма-цепь (IL2RG), такие как IL2, IL4, IL7, IL9, IL15 и IL21, важны для развития и функции B и Т-клеток [1]. Рецептор IL7 получил большое внимание, потому что он является маркером ранних лимфоидных клеток-предшественников, он необходим как для развития B, так и для Т-клеток, и недавно он был идентифицирован как доминирующий онкоген при острой лимфобластной лейкемии (ALL). Мы сосредоточимся здесь на рецепторном комплексе IL7 в качестве примера того, как сигнализация цитокинового рецептора может быть захвачена клетками лейкоза.
Рецептор IL7 представляет собой гетеродимерный рецептор, состоящий из альфа-цепи рецептора IL7 (IL7R) и общей гамма-цепи (IL2RG). Эти рецепторные единицы ассоциированы с Janus kinase 1 (JAK1) и JAK3 соответственно, и эти киназы активируются при связывании IL7 с рецептором (фиг.1). Мутации с потерями функции в IL2RG, IL7R или JAK3 приводят к нарушению развития B и T-клеток и выявлены у пациентов с тяжелой комбинированной иммунодефицитной болезнью, что ясно показывает, что эта сигнальная ось необходима для нормального развития лимфоцитов [1]. Напротив, мутации с усилением функции в IL7R, JAK1 или JAK3 приводят к лиганд-независимой активации передачи сигналов IL-1 и идентифицированы во ВСЕХ и в различных типах лимфомы. 1Симматическое представление комплекса сигнализации рецептора интерлейкина-7 (IL7). Стрелки указывают местоположения, которые часто оказываются мутированными. Активация рецепторного комплекса связыванием лиганда или мутацией в IL7R, JAK1 или JAK3 вызывает фосфорилирование белков STAT, что приводит к активации путей выживания и пролиферации
Активация мутаций в IL7R часто встречается в T-клетке ALL (T-ALL) и B-клетке ALL (B-ALL) [2, 3]. Эти IL7R-активирующие мутации расположены в экзоне 6 и в основном приводят к включению неспаренного цистеина, близкого к трансмембранному домену рецептора (фиг.1). Таким образом, мутантный белок IL7R может образовывать гомодимеры путем образования дисульфидных связей, что приводит к не зависящей от цитокина активации путей передачи сигналов ниже по течению. В меньшем количестве случаев ALL мутации IL7R не связаны с введением аминокислоты цистеина, и эти вставки происходят в трансмембранном домене, что, скорее всего, приводит к лиганд-независимой активации гетеродимерных рецепторов.
В дополнение к мутациям в самом рецепторе мутации в тирозинкиназе JAK3 часто встречаются у T-ALL [2, 4, 5], тогда как активирующие мутации JAK1 происходят на низкой частоте в T или B ALL (фиг.1) [ 2, 6]. Из-за ограниченной картины экспрессии IL7R мутации IL7R ограничены лимфоидными злокачественными новообразованиями, тогда как мутации JAK1 и JAK3 также можно ожидать при миелоидных лейкозах и даже при любом типе рака. Действительно, мутации JAK1 также были обнаружены во множестве эпителиальных опухолей с самой высокой частотой при гепатоцеллюлярной карциноме (http://cancer.sanger.ac.uk).
В течение последних десятилетий комбинированная химиотерапия была оптимизирована для лечения ВСЕХ, и детство ALL теперь можно вылечить более чем у 80% детей. Пациенты, однако, страдают серьезными краткосрочными и долгосрочными побочными эффектами интенсивного лечения, а взрослые ВСЕГДА имеют плохой результат. С увеличением понимания молекулярных дефектов, участвующих в патогенезе ВС, теперь можно разработать специфические для пациента методы лечения, в которых лечение основано на мутационном статусе лейкемических клеток. Поскольку рецепторный комплекс IL7 (JAK1, JAK3, IL7R) мутирован в до 25% случаев T-ALL, это может быть одной из новых терапевтических целей, которые необходимо изучить.
Белки тирозинкиназы представляют собой интересные белки с терапевтической точки зрения, поскольку эти ферменты легко мигрируют с помощью ингибиторов малых молекул, и эти белки часто мутируются и конститутивно активируются при раке. Ингибитор ABL imatinib произвел революцию в лечении хронического миелоидного лейкоза, а также в BCR-ABL-положительном ALL, сочетание ингибиторов ABL-киназы с химиотерапией показало многообещающие результаты. Поскольку начальные успехи с иматинибом были разработаны многие другие ингибиторы киназы, включая различные ингибиторы JAK-киназы (таблица 1). Хотя большинство из этих ингибиторов все еще находятся в стадии разработки, JAK1 / JAK2-селективный ингибитор ruxolitinib уже одобрен FDA для лечения пациентов с миелофиброзом, а JAK3-селективный ингибитор tofacitinib получил одобрение FDA для лечения пациентов с ревматоидным артритом. Эти данные показывают, что ингибиторы JAK-киназы можно безопасно вводить и открывать новые возможности для лечения T-ALL с мутациями IL7R, JAK1 или JAK3. С T-ALL, являющимся редким лейкозом, очень повезло, что многочисленные ингибиторы JAK уже доступны и могут быть потенциально использованы для лечения T-ALL.Table 1JAK1 и JAK3-селективных ингибиторов, которые в настоящее время находятся в клинических исследованиях
Сообщалось, что мутанты JAK1 [7], IL7R [3] и JAK3 [8] чувствительны к JAK-селективному ингибированию. JAK1 незаменим для мутантов IL7R, чтобы поддерживать активацию последующих белков, таких как STAT5 [3]. Аналогично, мы сообщили о нескольких строках доказательств того, что JAK1 необходим для трансформационных механизмов большинства JAK3-мутантов. Таким образом, хотя число ВСЕХ пациентов с определенными мутациями в IL7R, JAK1 или JAK3 невелико, все эти случаи вместе составляют около 27% случаев T-ALL и, вероятно, будут реагировать на ингибиторы JAK [9].
Недавно мы показали, что большинство мутаций JAK3, идентифицированных в образцах пациентов T-ALL, вызвали лейкемию в модели мыши [8]. В развитии мышиных моделей, экспрессирующих мутации JAK3, мы в основном фокусировались на мутации JAK3 M511I, которая является наиболее распространенной мутацией, обнаруженной в T-ALL. Мыши, получавшие трансплантацию костного мозга клетками, экспрессирующими мутант JAK3 M511I, развивали лимфопролиферативное заболевание в течение первых 12 недель с последующим прогрессированием в острую фазу, характеризующуюся быстрым увеличением количества лейкоцитов (WBC). Все животные в конечном итоге поддались болезни в течение 14-28 недель после приема трансплантации костного мозга [8]. Болезнь характеризовалась спленомегалией, увеличенным тимусом и увеличенными лимфатическими узлами. Все мыши продемонстрировали накопление CD8-положительных незрелых Т-клеток в периферической крови и кроветворных тканях. Лейкемические клетки трансплантировали вторичным реципиентным мышам и характеризовались наличием дополнительных мутаций в Notch2, Pten, Kras и других генах. Мыши, получившие трансплантацию костного мозга клеток, экспрессирующих JAK3 дикого типа, не развивали фенотипа болезни [8].
Первичные и вторичные трансплантированные мыши впоследствии использовали для проверки эффективности JAK3-селективного ингибитора, tofacitinib (Xeljanz, Pfizer), для лечения прогрессирования лейкемии. Мыши, получавшие тофацитиниб, показали снижение количества лейкоцитов, тогда как мыши, получавшие лечение плацебо, увеличивали количество WBC во время лечения. Патологический анализ тканей показал высокий процент апоптотических клеток у обработанных тофацитибом мышей, тогда как мыши, получавшие плацебо, имели очень малое количество апоптозных клеток. Масса селезенки и тимуса была значительно ниже у обработанных tofacitinib мышей по сравнению с мышами, получавшими плацебо. Эти данные показывают, что ингибиторы JAK, такие как тофацитиниб, проявляют активность в моделях лейкемии у мышей [8]. Однако, когда лечение прекращалось, количество лейкоцитов увеличивалось, и все животные в конечном итоге поддались болезни, показывая, что только ингибиторы киназы не могут привести к полному уничтожению лейкемических клеток в этой модели мыши.
В отдельном исследовании Мод и его коллеги исследовали эффективность ruxolitinib для лечения предшественника предшественников T-клеток ALL (ETP-ALL) с использованием образцов ксенотрансплантата человеческого лейкоза. Инъекция образцов ETP-ALL у иммунодефицитных мышей NSG приводила к расширению клеток лейкемии in vivo, что наблюдалось увеличением числа человеческих взрывных клеток в периферической крови и селезенке мышей NSG с течением времени. Лечение этих животных с помощью ruxolitinib, ингибитора JAK1 / JAK2, вызвало резкое сокращение периферических и селезенки, даже в качестве одного агента. Интересно отметить, что эффективность ruxolitinib наблюдалась не только в трех образцах с мутацией JAK1 или JAK3, но и в двух образцах ETP-ALL без мутации JAK1, JAK3 или IL7R [10]. Эти данные свидетельствуют о том, что ингибиторы JAK являются перспективными агентами для лечения T-ALL и что клинические испытания для проверки этих агентов оправданы. Мутации JAK1, JAK3 или IL7R предсказывают реакцию на ингибиторы JAK, но даже случаи T-ALL без таких мутаций могут потенциально реагировать на ингибирование JAK, скорее всего, из-за наличия других неизвестных механизмов, приводящих к активации JAK / STAT путь.
Целевые агенты, такие как ингибиторы тирозинкиназы, очень активны и специфичны и, как правило, проявляют сильные противораковые эффекты, но страдают от того факта, что они связываются с целевой киназой в одной конкретной части, обычно в АТФ-связывающем кармане. Как следствие, онкогенные киназы могут стать устойчивыми к ингибитору путем мутации одной аминокислоты, что было обнаружено почти во всех клинических применениях целенаправленной терапии рака.
Пока еще рано говорить о том, будут ли ингибиторы JAK проявлять клиническую эффективность для лечения T-ALL, но если это будет успешным, можно ли ожидать сопротивления и как мы можем разработать лучшие ингибиторы? Можно ли разработать стратегию предотвращения или ограничения развития сопротивления? Интимное взаимодействие между киназой JAK1 и JAK3 у рецептора IL7 может предложить возможности для предотвращения развития резистентности. Недавнее исследование показало, что клетки, трансформированные JAK3-мутантом, могут стать устойчивыми к JAK3-селективному ингибитору, приобретя еще одну активирующую мутацию в JAK1 [11]. Ориентируясь на JAK1 и JAK3 в клетках мутантного лейкоза JAK3, клеткам лейкемии может быть очень сложно преодолеть этот блок только одной мутацией. Более того, наше исследование показало, что комбинация JAK3-селективных и JAK1-селективных ингибиторов имела явный синергетический эффект на рост мутантных клеток JAK3 [8]. Таким образом, доза обоих лекарств может быть снижена при одновременном достижении хорошего ингибирования сигнальных путей.
Хотя комбинированное ингибирование как JAK1, так и JAK3 может быть полезным для лечения подмножества случаев T-ALL, также ясно, что другая группа мутантов очень сильно зависит от JAK1 или JAK3. Будет важно дополнительно прояснить механизмы, с помощью которых различные мутанты JAK1, JAK3 и IL7R трансформируют клетки. Лучшее понимание точных путей передачи сигналов, которые активируются каждым из мутантов, может помочь рациональному выбору целевых агентов для достижения лучших ответов и предотвращения развития резистентности.
острый лимфобластный лейкоз
В-клеточный острый лимфобластный лейкоз
ранний тимус-предшественник острый лимфобластный лейкоз
преобразователь сигнала и активатор транскрипции
Т-клеточный острый лимфобластный лейкоз
лейкоцит
Конкурирующие интересы
Авторы заявляют, что у них нет конкурирующих интересов.
Вклад авторов
SD и JC написал рукопись. Оба автора прочли и утвердили окончательную рукопись.
Авторы выражают благодарность somersault18: 24 за подготовку фигуры.
rupubmed.com
лекарство на миллиард долларов — ЦВТ ХимРар
«Янссен» (Janssen), принадлежащая «Джонсон энд Джонсон» (Johnson & Johnson), и «Тераванс байофарма» (Theravance Biopharma) договорились о совместной разработке экспериментального TD-1473 последней, предназначенного для терапии воспалительных заболеваний кишечника, включая неспецифический язвенный колит и болезнь Крона.

Соответствующие клинические испытания фаз IIb/III и II будут запущены в текущем году. «Тераванс» получит 100 млн долларов авансом и еще до 900 млн долларов по мере прохождения проектом определенных стадий развития, а также двузначное роялти от продаж готового препарата за пределами США.
TD-1473, будучи пероральным ингибитором Янус-киназ (JAK) всех подтипов (JAK1, JAK2, JAK3, TYK2), работает, в отличие от любых других разрабатываемых против воспалительных заболеваний кишечника JAK-ингибиторов, прямиком в месте воспаления кишечной стенки. Другими словами, заявлено о минимизировании системного воздействия. Ингибирование Янус-киназ отражается на сигнальном пути JAK-STAT в виде модулирования активности широкого спектра провоспалительных цитокинов.
Вместе с тем велика проблематика негативных побочных эффектов, наблюдающихся при системном воздействии JAK-ингибиторов, включая тромбоэмболические события, инфекции, перфорацию желудка и кишечника, нейтропению, раковые заболевания. Именно потому Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) отказалось одобрять «Олюмиант» (Olumiant, барицитиниб), пока доступный только в Европе.
В случае «Тераванс» и ее TD-1473 указанные неприятности, похоже, удалось обойти. Другими словами, отсутствует ограничение назначаемой дозы препарата, и он сможет продемонстрировать свою эффективность на полную катушку. В клинических исследованиях фазы Ib было засвидетельствовано минимальное системное воздействие молекулы, которая не привела к развитию умеренно-тяжелых негативных реакций, ассоциированных с системной иммуносупрессией.
Сейчас на рынке представлены три препарата этого класса: «Зелджанс»/«Яквинус» (Xeljanz/Jakvinus, тофацитиниб), «Джакафи»/«Джакави» (Jakafi/Jakavi, руксолитиниб) и «Олюмиант» — они ингибируют соответственно JAK1/3, JAK1/2 и JAK1/2. Медикаменты продвигают «Пфайзер» (Pfizer), «Инсайт» (Incyte)/«Новартис» (Novartis) и «Илай Лилли» (Eli Lilly)/«Инсайт». В настоящее время они разрешены в терапии ревматоидного артрита/псориатического артрита, миелофиброза/истинной полицитемии, ревматоидного артрита. Разумеется, проводится множество исследований, призванных расширить спектр назначений.
Другие фармкомпании продолжают доводить до ума свои JAK-ингибиторы. Так, «ЭббВи» (AbbVie) уверена в светлом будущем JAK1-ингибитора упадацитиниба (upadacitinib), который уже продемонстрировал впечатляющие результаты в терапии ревматоидного артрита, болезни Крона, атопического дерматита (экземы), неспецифического язвенного колита, псориатического артрита.
JAK-ингибиторы видятся более чем прибыльным направлением деятельности. Уместно напомнить, что в январе «Селджен» (Celgene) купила «Импакт байомедисинз» (Impact Biomedicines) за общую сумму в 7 млрд долларов, включая авансовых 1,1 млрд долларов. Взамен был получен готовый к регистрации федратиниб (fedratinib), высокоизбирательный JAK2-ингибитор, пригодный для лечения трех основных типов миелопролиферативных новообразований: первичного миелофиброза (в том числе рефрактерного к руксолитинибу), истинной полицитемии, эссенциальной тромбоцитемии.
Источник: https://mosmedpreparaty.ru/
chemrar.ru
Симптомы комбинированных Т и В-клеточных иммунодефицитов
У человека тяжелая комбинированная иммунная недостаточность впервые описана в 1950 г. в Швейцарии у нескольких младенцев с лимфопенией, умиравших от инфекций в течение первого года жизни. Именно поэтому в течение многих лет в литературе встречалось выражение «швейцарский тип ТКИН». В последующие годы было выявлено, что тяжелая комбинированная иммунная недостаточность включает множество синдромов, имеющих различную генетическую природу и различные типы наследования (X-сцепленное в 46% случаев и аутосомно-рецессивное в 54%). Общая частота ТКИН — 1:50 000 новорожденных. В настоящее время известна генетическая природа примерно 15 форм ТКИН, которые, на основании различий в иммунологическом фенотипе, могут быть разделены на 5 групп: T-B+ NK+, T-B- NK+,T-B+ NK-,T+B+NK- и Т-B-NK-.
Основные клинические проявления тяжелой комбинированной иммунной недостаточности практически не зависят от генетического дефекта. Для больных с ТКИН характерно раннее, в первые недели и месяцы жизни, начало клинических проявлений заболевания в виде гипоплазии лимфоидной ткани, упорной диареи, мальабсорбции, инфекций кожи и слизистых, прогрессирующего поражения респираторного тракта. Возбудителями инфекций являются бактерии, вирусы грибы, условно-патогенные микроорганизмы (в первую очередь Pneumocyctis carini). Цитомегаловирусная инфекция протекает в виде интерстициальной пневмонии, гепатита, энтеровирусы и аденовирус вызывают менингоэнцефалиты. Очень часть встречается кандидоз слизистых и кожи, онихомикоз. Характерно развитие регионарной и/или и генерализованной БЦЖ инфекции после вакцинации. На фоне тяжелых инфекций наблюдается отставание в физическом и моторном развитии. Следует помнить, что даже при наличии тяжелой комбинированной иммунной недостаточности у младенцев не сразу развиваются все вышеперечисленные симптомы, и в течение 2-3 месяцев они могут расти и развиваться почти нормально, особенно, если вакцинация БЦЖ не была сделана. Трансплацентарная передача материнских лимфоцитов может вызвать симптомы реакции «трансплантат против хозяина» (РТПХ), называемой в этом случае материнско-фетальной РТПХ. Она проявляется в основном а виде кожной эритематозной или папулезной сыпи и поражения печени.
При лабораторном обследовании в большинстве случаев выявляется выраженная лимфопения, гипогаммаглобулинемия и снижение пролиферативной активности лимфоцитов. Близкое к норме количество лимфоцитов может являться результатом трансплацентарной передачи лимфоцитов от матери. Как было отмечено выше, Т-лимфоциты значительно снижены при всех формах тяжелой комбинированной иммунной недостаточности, однако число и функции В лимфоцитов и NK клеток зависит от генетического дефекта, лежащего в основе ТКИН. В редких случаях отмечается нормальная концентрация иммуноглобулинов, однако их неадекватная специфичность ведет к полной неэффективности гуморального звена. Далее мы рассмотрим некоторые особенности патогенеза различных форм тяжелой комбинированной иммунной недостаточности.
Молекулярно-генетические особенности различных форм тяжелой комбинированной иммунной недостаточности
Т- В- NK- ТКИН
- Ретикулярная дисгенезия
Ретикулярная дисгенезия является редкой формой тяжелой комбинированной иммунной недостаточности, характеризующейся нарушением созревания лимфоидных и миелоидных предшественников на ранних этапах развития в костном мозге. Предполагается аутосомно-рецессивное наследование, однако в связи с редкостью заболевание оно не доказано. Молекулярно-генетические основы заболевания не известны. Заболевание характеризуется выраженной лимфопенией, гранулоцитопенией, тромбоцитопенией, тяжелыми инфекциями, проводящими к ранней гибели пациентов.
T- B+ NK- ТКИН
- Х-сцепленная тяжелая комбинированная иммунная недостаточность
Х-сцепленная ТКИН, или дефицит g цепи — наиболее часто встречающаяся форма (более 50% всех форм тяжелой комбинированной иммунной недостаточности). Она развивается в результате мутации гена общей у цепи (CD132) рецепторов интерлейкинов 2, 4, 7, 9,15. Мутация у цепи приводит к блокаде рецепторов, в результате чего клетки-мишени не способны ответить на действие соответствующих интерлейкинов. Иммунологические нарушения, развивающиеся у этих больных, характеризуются отсутствием Т-клеток и NK клеток и повышением количества В-клеток. В результате отсутствия T-клеточной регуляции продукция иммуноглобулинов В-клетками резко снижена.
Тирозинкиназа семейства Janus — Jak3 необходима для передачи активационного сигнала от общей уцепи IL2, 4, 7, 9, 15 к ядру клетки. Дефицит jak3 вызывает такие же глубокие нарушения дифференцировки Т- и NK-клеток, как дефицит общей уцепи. Иммунологические нарушения и клинические проявления у больных с дефицитом Jak3 аналогичны таковым при X-сцепленной ТКИН.
Трансмембранная протеинтирозинкиназа CD45, специфичная для гемопоэтических клеток, необходима для передачи сигнала с антигенного рецептора T- и В-клеток. Мутации гена CD45 приводят к развитию ТКИН, характеризующейся резким снижение количества T клеток, нормальным содержанием В-клеток и прогрессирующим снижением концентраций сывороточных иммуноглобулинов. Число NK лимфоцитов снижено, однако не полностью.
Т- В- NK+ ТКИН
- Полный дефицит RAG1/RAG2
Белковые продукты активирующих рекомбинацию генов — recombination activating genes (RAG1 и RAG2) инициируют формирование иммуноглобулинов и T-клеточных рецепторов, необходимых для дифференцирован В- и T-клеток. Таким образом, мутации RAG генов приводят к формированию тяжелой комбинированной иммунной недостаточности. При этой форме иммунодефицита отсутствуют T- и В-клетки, в то время как количество NK-клеток нормально. Количество сывороточных иммуноглобулинов резко снижено.
- Радиочувствительная ТКИН (дефицит Artemis)
В 1998 году были идентифицированы пациенты с Т-B-NK+ тяжелой комбинированной иммунной недостаточности, не имеющие мутаций генов RAG1/RAG, отличающиеся высокой чувствительностью к ионизирующей радиации и имеющие нарушения восстановление двухцепочечных разрывов ДНК (DNA double strand break repair), T- и В-лимфоциты распознают антигены с помощью молекул Т-клеточных рецепторов (TCR) и иммуноглобулиноз. Антиген-специфичные участки этих рецепторов состоят из трех сегментов: V- (вариабельных), D (разнообразия) и J (объединения). Полиморфизм антиген-специфичных участков TCR и иммуноглобулинов обеспечивается процессом соматической реарранжировки и V(D)J рекомбинации. В процессе рекомбинации генов иммуноглобулинов и TCR RAG протеины индуцируют двухцепочечные разрывы ДНК. Восстановление индуцированных радиацией и спонтанных разрывов ДНК требует участия ряда протеинкиназ и недавно идентифицированного фактора, названного Artemis. Artemis необходим для остановки клеточного цикла в случае повреждения ДНК.
Мутации гена Artemis приводят к развитию аутосомно-рецессивной тяжелой комбинированной иммунной недостаточности с повышенной радиочувствительностью, характеризующейся отсутствием Т- и В-лимфоцитов и хромосомной нестабильностью. Отличительной чертой клинических проявлений, помимо характерных для scex ТКИН, является наличие номоподобных поражений слизистых рта и других локализаций.
T- B+ NK+ TKИH
Предшественники T- и В-клеток экспрессируют функциональный IL7R, состоящий из а цепи и общей у цепи. Экспрессия этого рецептора критична для созревания T-лимфоцитов, но не критична для развития В-лимфоцитов. Мутации гена альфа цепи IL-7R приводит к развитию ТКИН, с фенотипом T-B-NK+ и выраженным снижением концентраций сывороточных; иммуноглобулинов.
T+ B+ NK- ТКИН
В 2001 году, впервые, Gilmour KC et al. описали пациента с низким абсолютным количеством T-лимфоцитов, нормальным количеством В-клеток и полным отсутствием NK-клеток. И хотя никаких мутаций в генах общей у цепи или JAK3 обнаружено не было, функциональные исследования показали нарушение фосфорилирования JAK3 через комплекс IL2R. Последующий цитометрический анализ показал значительное снижение экспрессии beta цепи рецептора IL15 (IL15Rbeta). Однако, мутации гена IL15Rbeta выявить не удалось, что заставляет предполагать наличие дефектов транскрипции, которые оказались ответственными за отсутствие экспрессии цепи IL15Rbeta.
- Дефицит ферментов пуринового обмена
Дефицит двух ферментов, катализирующих пуриновый метаболизм — аденозиндезаминазы (ADA) и пуриннуклеозидфосфарилазы (PNP), ассоциирован с развитием комбинированной иммунной недостаточности. Вследствие отсутствия этих ферментов накапливаются токсичные для клеток продукты — деэоксиаденозин и дезоксигуанозин, которые частично фосфорилируются в лимфоидных клетках, превращаясь в соответствующие дезоксинуклеозидтрифосфаты. Токсичность этих продуктов особенно важна в быстро делящихся клетках и заключается в ингибировании синтеза ДНК, индукции апоптоза, нарушении метилирования и др. Оба эти состояния гетерогенны по клиническим проявлениям в зависимости от локализации мутации на протяжении генов итого, насколько в результате страдает функция соответствующего фермента.
- Дефицит аденозиндезаминазы (ADA)
Дефицит аденозиндезаминазы — одна из первых идентифицированных форм ТКИН. Ген аденозиндезаминазы находится на 20ql3.ll. Известно более 50 вариантов мутаций гена ADA. Существует зависимость между генетически детерминированной резидуальной активностью аденозиндезаминазы и метаболическим и клиническим фенотипом. ADA экспрессируется в различных тканях, особенно высока его экспрессия в незрелых тимоцитах и В лимфоцитах, по мере созревания клеток экспрессия АДА уменьшается. При дефиците аденозиндезаминазы в клетках накапливаются дезоксиаденозинтрифосфат и S-аденозилгомоцистеин. Эти метаболиты ингибируют пролиферацию ТT- и В-лимфоцитов.
У большинства пациентов с дефицитом аденозиндезаминазы в раннем возрасте проявляются все признаки ТКИН. Это, как правило, больные с самым низким число лимфоцитов и самыми ранними и тяжелыми проявлениями. У этих больных не отмечается приживление материнских лимфоцитов. Кроме иммунологических, нарушение пуринового обмена может вызывать скелетные нарушения. Так, при рентгенологическом исследовании выявляются увеличенные косто-хондральные сочленения (как при рахите), расширение концов ребер, тазовая дисплазия. У больных также описаны следующие неврологические изменения: нистагм, сенсорная глухота, спастические нарушения, нарушение психомоторного развития (независимо от инфекций). Частым признаком дефицита аденозиндезаминазы является повышение трансаминаз, вероятно свидетельствующее о наличии токсического гепатита.
В последние годы описаны варианты с «поздним началом» ADA-дефицита и выявлены даже здоровые индивидуумы с частичным дефицитом фермента.
Ведение больных тяжелыми проявлениями ADA-дефицита практически не отличается от терапии других ТКИН. Однако экспериментальным методом является назначение заместительной внутримышечной терапии ферментом PEG-ADA в дозе 15-30 мг/кг/неделю. Коррекция дефектов требует длительного и постоянного лечения. Число и функция Т лимфоцитов как правило улучшается к 6-12 недели терапии, однако даже после длительного лечение (10 лет) у большинства больных сохраняется лимфопения и нарушение митогенного ответа.
- Дефицит пурин-нуклеозндфосфорилазы (PNP)
Ген PNP расположен на 14ql3. В отличие от ADA, активность пурин-нуклеозндфосфорилазы увеличивается по мере созревания Т лимфоцитов. При дефиците PNP в клетках накапливается деэоксигуанозинтрифосфат, ингибирующий пролиферацию Т-лимфоцитов.
Также как и при дефиците аденозиндезаминазы, у большинства пациентов с дефицитом пурин-нуклеозндфосфорилазы клинические проявления ТКИН развиваются в младенческом возрасте, хотя в отдельных случаях описано более позднее начало. Сопутствующими синдромами при дефиците PNP являются урикемия и урикурия. Часто у больных с дефицитом пурин-нуклеозндфосфорилазы отмечаются аутоиммунные (гемолитическая анемия, тромбоцитопения, нейтроления, системная красная волчанка) и неврологические (плегии, парезы, атаксия, тремор, задержка умственного развития) проявления. У больных отмечается повышенная склонность к онкологическим заболеваниям. При лабораторном исследовании отмечается резкое снижение T лимфоцитов и как правило нормальное число В лимфоцитов. Проявлением дисрегуляции В лимфоцитов являются повышение уровня иммуноглобулинов, гаммапатия, наличие аутоантител.
Синдром «голых лимфоцитов» является врожденным иммунодефицитом, развивающимся из-за отсутствия на поверхности клеток экспрессии молекул II класса главного комплекса гистосовместимости (МНС II). При этом заболевании из-за дефектов генов, контролирующих МНС II, не происходит экспрессии его молекул, необходимых для дифференцировки и активации CD4+клеток, нарушается селекция T-клеток в тимусе, и развивается тяжелый иммунодефицит. Поврежденные гены кодируют четыре высокоспецифичных транскрипционных фактора (RFXANK, RFX5, RFXAP и СИТА), регулирующих экспрессию МНС II. Первые три являются субъединицами RFX (Regulatory Factor X) — тримерного, присоединяющего ДНК комплекса, который регулирует все промоторы МНС II. CIITA {Class II Trans activator) является не связывающим ДНК ко-активатором, который контролирует экспрессию МНС II.
Заболевание характеризуется типичными клиническими признаками ТКИН, которые, однако, протекают легче. Так, а группе 9 нетрансплантированных больных с этим заболеванием средняя продолжительность жизни составила 7 лет.
При лабораторном исследовании отмечается значительное снижение CD4+ лимфоцитов, при как правило нормальном числе CD8+ лимфоцитов. У некоторых больных отмечается отсутствие экспрессии не только молекул МНС II, но и МНС I. В целом отмечается выраженная недостаточность T клеточного ответа, продукция иммуноглобулинов также резко снижена.
ТАР {Transporter Associated Protein) необходим для транспорта антигенных пептидов в эндоллазматический ретикулум и присоединения их к молекулам МНС I класса. Выявлены дефекты 1 и 2 субъединиц ТАР (ТАР1 и ТАР2). Характерными лабораторными проявлениями у пациентов с дефицитом ТАР являются: отсутствие экспрессии МНС I класса, близкие к нормальным уровни иммуноглобулинов (у некоторых больных отмечены селективный дефицит IgM), отсутствие антительного ответа на полисахаридные антигены. У различных больных отмечалось нормальное или прогрессивно снижающееся число CD8 Т лимфоцитов, остальные субпопуляции лимфоцитов как правило были нормальными. При этой форме КИН существует высокая чувствительность к бактериальным инфекциям слизистых респираторного тракта, характерны гранулематозные поражения кожи. Вирусные инфекции и инфекции, вызванные внутриклеточными патогенами, встречаются редко. У отдельных пациентов описано бессимптомное течение и позднее начало клинических проявлений иммунодефицита.
Мутации гена альфа-цепи рецептора IL-2 (IL2Rct) {CD25) приводят к развитию КИН с снижением количества и нарушением пролиферации периферических T-клеток и нормальным развитием В-клеток. Дифференцировка тимоцитов не нарушена, но, несмотря на нормальную экспрессию CD2, CD3, CD4 и CD8, CD25, кортикальные тимоциты не экспрессируют CD1. Больные имеют повышенную чувствительность к вирусным инфекциям (ЦМВ и др.), также с раннего возраста страдают повторными бактериальными и грибковыми инфекциями, хронической диареей, У больных также отмечается лимфопролиферация, сходной с таковой при АЛПС. Предполагается, что в основе ее лежит нарушение регуляция апоптоза в тимусе, приводящая к экспансии аутореактивных клонов в различных тканях.
- Дефицит СВЗу и CD3е
Антиген-распознающий рецепторный комплекс T-клеток состоит из собственно T-клеточного рецептора (TCR) и молекулы CD3. Существует два типа TCR, каждый из которых состоит из двух пептидных цепей — аb и уv. Основной функцией- TCR является связывание антигенного пептида, ассоциированного с продуктами главного комплекса гистосовместимости, a CD3 — передача антигенного сигнала в клетку. CD3 включает молекулы 4-5 типов. Все цепи комплекса CD3 (у, v, e, £, t) являются трансмембранными белками. Мутации генов цепей у, v или £ приводят к снижению количества зрелых T-клеток с низкой экспрессией TCR. Мутации гена e цепи приводит к нарушению дифференцировки тимоцитов на уровне CD4-CD8-. У человека дефицит СD3 у приводит к снижению количества CD8+ Т-лимфоцитов и CD4+CD45RA+, содержание CD4+CD45R0+, В- и NК-клеток и концентрации сывороточных иммуноглобулинов — нормальны. Клинический фенотип при дефиците CD3y и CD3e варьирует даже среди членов одной семьи от проявлений до довольно мягкого течения заболевания.
Протеин-тирозинкиназы семейства ZAP70/Syk играют важную роль в передаче сигнала от антиген-распознающего рецептора, они необходимы для нормального развития Т-лимфоцитов. ZAP70 необходима для дифференцировки ab Т-лимфоцитов. При дефиците ZAP70 развивается селективный дефицит CD8+ клеток. Количество CD4+ циркулирующих клеток нормально, но они имеют выраженные нарушения функций в виде отсутствия продукции IL-2 и пролиферативной активности. Концентрации сывороточных иммуноглобулинов снижены.
ilive.com.ua
Мутация в гене JAK2
01.05.2017 22:22:04
11012
Что такое мутация JAK2?
Ген JAK2 расположен на 9-й хромосоме (в положении 9p24) и кодирует протеиновую тирозинкиназу JAK2, которая, в свою очередь входит в группу янусовых протеинкиназ (JAK1, LAK2, JAK3, JAK4).Гены JAK1 и JAK2 отвечают за продукцию и формирование STAT (signal transducers and activators of transcription) — сигнала активации деления клетки. Белки STAT, в свою очередь, отвечают за транскрипцию тех или иных генов в клетке.
Ген JAK2 представлен во всех клетках тела, в том числе и клетках крови, где принимает участие в передаче сигнала, активируя транскрипцию во многих клеточных процессах.Мутация JAK2 приводит к дерегуляции JAK-STAT и последующему аномальному накоплению онкопротеинов с фенотипическими чертами миелопролиферативных неоплазий.

Мутация в гене JAK2 присутствует при следующих заболеваниях:
- истинная полицитемия 99%;
- эссенциальная тромбоцитемия 55%;
- идиопатический миелофиброз 65%;
- хронический миелолейкоз;
- миелодиспластический синдром с кольцевидными сидеробластами.
Появление мутации в гене JAK2 нарушает равновесие в процессе выработки клеток крови, что приводит к появлению у человека таких хронических миелопролиферативных заболеваний (ХМПЗ) как: истинная полицитемия, эссенциальная тромбоцитемия и идиопатический миелофиброз.Хронические миелопролиферативные заболевания (ХМПЗ), без должной терапии, могут перерасти в постполицитемический и посттромбоцитемический миелофиброз, а уже они обычно перерождаются в острый миелобластный лейкоз.
Определение мутации генов JAK1 и JAK2 позволяет выявить предрасположенность к развитию хронических миелопролиферативных заболеваний. Также, определение мутаций в гене JAK1 и JAK2 показано пациентам, которым поставлен диагноз миелобластный лейкоз. Необходимо помнить, что своевременное лечение хронических миелопролиферативных заболеваний снижает риск развития острого миелобластного лейкоза.
У пациентов с выявленными мутациями генов JAK1-2, при миелофиброзе, показано применение таргетного препарата Jakavi (Руксолитиниб). Он доказал свою высокую эффективность в нескольких клинических исследованиях и успешно применяется в современной онкологии для лечения пациентов с мутациями с гене JAK1 и JAK 2.
Подпишитесь на нашу еженедельную рассылку
worldofoncology.com
Ингибитор JANUS-киназы • СЛИПАПС
В прошлом году в России закончился набор в два исследования с пациентами с ревматоидным артритом. К сожалению, я их проморгал и «радарных» постов о них не было. Оба исследования спонсировала компания AbbVie, целью исследований было изучение эффективности и безопасности препарата ABT-494, ингибитора киназы JAK1.
В этом посте я немного расскажу о том, что показали эти исследования и чего нам ждать дальше.
ABT-494 это низкомолекулярный селективной ингибитор JAK1-киназы второго поколения, для применения per os. Пока что AbbVie разрабатывает это лекарство для пациентов с ревматоидным артритом, псориазом и болезнью Крона.
Ревматоидный артрит (РА) – это аутоиммунное заболевание со сложным патогенезом и обусловленной целым комплексом причин и провоцирующих факторов. Пока что оно считается неизлечимым, и лучшее, что может дать пациенту современная медицина – это длительная ремиссия.
Самый перспективный класс препаратов для лечения РА и других аутоиммунных заболеваний – это моноклональные антитела, подавляющие эффекты про-воспалительных цитокинов и, таким образом, снижающие интенсивность воспаления.
Таких антител пока немного, это:
- Антитела против TNFα – Инфликсимаб, Адалимумаб, Голумумаб и Цетролизумаб
- Антитела против В-лимфоцитов – Ритуксимаб
- Антитела против рецептора интерлейкина-6 – Тоцилизумаб
Помимо них есть еще растворимые рецепторы, перехватывающие цитокины и не дающие им работать. К сожалению, эффективность этих препаратов ограничена, и у части пациентов они не позволяют добиться клинически значимой ремиссии. Тем не менее, эффект у них есть, а значит мишени выбраны правильно, просто биология клетки настолько сложна, что надежно воздействовать на все механизмы, участвующие в заболевании невозможно.
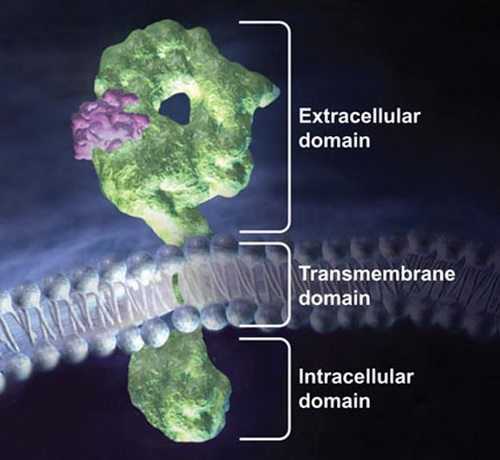
Цитокины вызывают биологические эффекты в результате воздействия на свои рецепторы, расположенные на мембранах иммунных, эпителиальных и других клеток. Рецептор цитокина состоит из трех доменов: внеклеточного, трансмембранного и внутриклеточного –
Присоединение цитокина к внеклеточному домену запускает цепную реакцию, которая начинается во внутриклеточном домене. К каждому внутриклеточному домену присоединен неактивный фермент семейства JAK или JANUS-киназ. Слово JAK – это сокращение от Just Another Kinase, а JANUS-киназа – творческая переработка этой аббревиатуры. JAK семейство включает четыре фермента: JAK1, JAK2, JAK3 и TYK4.
Когда цитокин присоединяется к своему рецептору – фермент JAK активируется и фосфорилирует сам себя, затем тирозиновые основания рецептора (его внутриклеточного домена), а затем — транскрипционные факторы семейства Signal Transducers and Activation of Transcription (STATs).
В результате этого фосфорилированные (то есть активированные) STAT молекулы отправляются в ядро и повышают там экспрессию генов, кодирующих про-воспалительные молекулы.
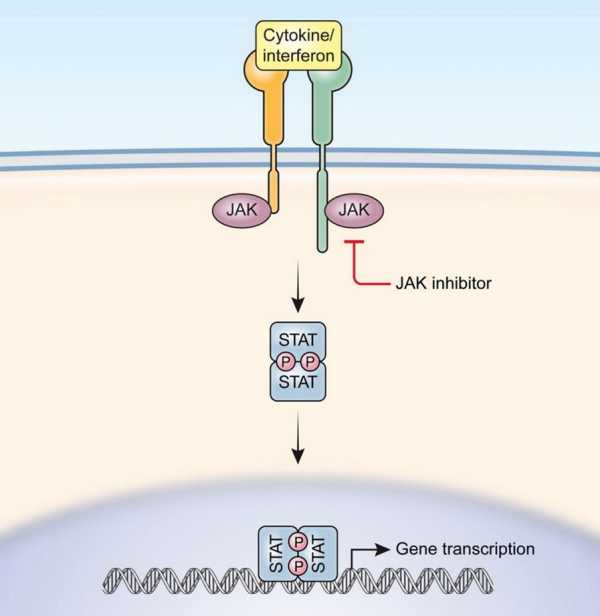
Когда моноклональное антитело (лекарство) блокирует про-воспалительный цитокин, оно действует снаружи клетки и всего лишь препятствует связыванию цитокина со своим рецептором, но проблема заключается в том, что при определенных обстоятельствах (например, при активирующих мутациях JANUS-киназ), каскад реакций внутри клетки может начать работать сам, и без цитокина. И тогда его блокирование антителом ничего не даст.
Представь, что цитокин — это ключ от замка зажигания автомобиля. Тогда цитокиновый рецептор — это замок, а все внутриклеточные процессы — это зажигание и двигатель. Обычно машину заводят ключом, и если ключ «заблокировать антителом», то завести не удастся. Только вот проблема в том, что иногда машина заводится и без ключа.
Поэтому нужен альтернативный подход – блокировать не ключ, а весь исполнительный механизм замка. И для этой цели неплохо подходит фермент JAK1. На рынке уже есть один ингибитор JAK1, одобренный для лечения пациентов с ревматоидным артритом – это Xeljanz® (Tofacitinib) производства Pfizer.

Кстати, обратите внимание, если препарат заканчивается на «mab» — это моноклональное антитело, а если на «nib» — это низкомолекулярный ингибитор тирозин-киназы. Как правило, так.
FDA одобрила Xeljanz® в 2012 году, для перорального применения у взрослых пациентов со средне-тяжелым ревматоидным артритом, которым не помогает метотрексат.
Что известно о ABT-494?
Это конкурент Xeljanz®.
В сентябре 2015 года AbbVie представила результаты двух плацебо-контролируемых исследований II фазы препарата ABT-494 – BALANCE I и BALANCE-II. ABT-494 – это селективный ингибитор JAK1. Исследования включали пациентов со средне-тяжелым ревматоидным артритом, которым не помогают анти-TNF препараты (BALANCE I) или метотрексат (BALANCE II).
Оба исследования показали впечатляющие результаты –
BALANCE I (4 разных дозы ABT-494 против плацебо на 276 пациентах):
Улучшение состояния по критериям ACR через 12 недель лечения:
- Как минимум на 20% 56% — 71% пациентов на ABT-494 против 35% на плацебо
- Как минимум на 50% 24% — 44% пациентов на ABT-494 против 17% на плацебо
- Как минимум на 70% 13% — 27% пациентов на ABT-494 против 4% на плацебо
BALANCE II (5 разных доз ABT-494 против плацебо на 300 пациентах):
Улучшение состояния по критериям ACR через 12 недель лечения:
- Как минимум на 20% 65% — 82% пациентов на ABT-494 против 50% на плацебо
- Как минимум на 50% 40% — 50% пациентов на ABT-494 против 20% на плацебо
- Как минимум на 70% 16% — 31% пациентов на ABT-494 против 7% на плацебо
Процент побочных эффектов, потребовавших прекратить прием препарата, составил < 5% в BALANCE I и < 3% BALANCE II.
Таких результатов было достаточно для того, чтобы принять решении о переходе программы в III фазу, о чем и AbbVie и объявил 8 января 2016 года. Всего будет пять регистрационных исследований III фазы, которые все вместе включат 4,000 пациентов с ревматоидным артритом.
Первое исследование (NCT02629159; SELECT-COMPARE) уже стартовало. Оно продлится до июля 2018 года и пока включило 58 центров в США, Австралии, Канаде, Новой Зеландии и Пуэрто Рико. Я проверил на сайте Минздрава – мы пока в этой программе не участвуем, но 4000 пациентов – это много, так что есть шанс, что хотя бы в одно исследование пригласят.
Буду проверять, и, если что – дам знать.
Показалось интересным или полезным — подпишитесь на анонсы новых статей в наших пабликах ВКонтакте и Фейсбуке.
slipups.ru
что это такое, лечение, прогноз, симптомы, стадии, признаки
Также может развиваться гепатоспленомегалия. Диагноз устанавливается на основании общего анализа крови, тестирования на наличие мутаций гена 1АК2 и клинических критериев. Лечение включает использование низких доз аспирина у всех пациентов и миелосуппрессивных препаратов у пациентов в группе высокого риска. Кровопускание раньше было стандартом лечения, но сейчас его роль противоречива.
Что такое истинная полицитемия
Истинная полицитемия — это наиболее распространенное миелопролиферативное заболевание. Заболеваемость им в США составляет 1,9/100 000, при этом риск увеличивается с возрастом. ИП несколько чаще возникает у мужчин. У детей ИП встречается очень редко.
Патофизиология истинной полицитемии
При ИП отмечается усиленная пролиферация всех клеточных ростков. В связи с этим ИП иногда называют панмиелозом из-за увеличения содержания представителей всех 3 клеточных линий периферической крови. Усиленная продукция одного эритроцитарного ростка называется эритроцитозом. Изолированный тромбоцитоз может наблюдаться при ИП, однако чаще он возникает по другим причинам (вторичный эритроцитоз).
Внекостномозговое кроветворение может происходить в селезенке, печени и других органах, которые могут служить местом образования клеток крови. Оборот клеток периферической крови возрастает. В конечном итоге заболевание может перейти в фазу истощения, проявления которой неотличимы от первичного миелофиброза. Трансформация в острый лейкоз отмечается редко, однако риск возрастает при использовании алкилирующих препаратов и радиоактивного фосфора. Последний следует использовать только в редких случаях или не использовать вовсе.
Осложнения. При ИП возрастает объем циркулирующей крови и увеличивается ее вязкость. Пациенты склонны к развитию тромбоза. Тромбоз может возникать в большинстве сосудов, приводя к инсультам, транзиторным ишемическим атакам или к синдрому Бадда-Киари. В прошлом эксперты считали, что повышенная вязкость крови является фактором риска тромбоза. Последние исследования указывают на то, что риск тромбоза может в первую очередь зависеть от выраженности лейкоцитоза. Однако эту гипотезу еще предстоит проверить в специально разработанных для этой цели проспективных исследованиях.
Функция тромбоцитов может быть нарушена, что повышает риск кровотечений. Ускоренный оборот клеток может вызывать повышение концентрации мочевой кислоты, тем самым увеличивая риск развития подагры и формирования камней в почках.
Генетические факторы. Клональный гемопоэз является отличительной чертой ИП. Это свидетельствует о том, что причиной пролиферации является мутация стволовых кроветворных клеток. Мутация JAK2 V617F (или одна из нескольких других более редких мутаций гена JAK2) обнаруживается практически у всех пациентов с ИП. Однако практически с полной уверенностью можно утверждать, что существуют и другие мутации, лежащие в основе заболевания. Они поддерживают JAK2 белок в состоянии постоянной активности, что приводит к избыточной пролиферации клеток независимо от концентрации эритропоэтина.
Признаки и симптомы истинной полицитемии
Её обнаруживают или случайно по высокому гемоглобину или по симптомам повышенной вязкости, таким как утомляемость, потеря концентрации внимания, головные боли, головокружение, потемнения в глазах, кожный зуд, носовые кровотечения. Иногда она проявляется заболеваниями периферических артерий или поражением сосудов головного мозга. Пациенты часто плеторичны, и у большинства пальпируют увеличенную селезёнку. Могут возникать тромбозы и часто пептическая язва, иногда осложнённая кровотечением.
Истинная полицитемия часто протекает бессимптомно. Иногда увеличение числа циркулирующих эритроцитов и повышение вязкости сопровождаются слабостью, дурнотой, нарушениями зрения, усталостью и одышкой. Частым симптомом является зуд, особенно после душа. Может отмечаться покраснение лица и расширение вен сетчатки, а также покраснение и болезненность ладоней и подошв, иногда в сочетании с ишемией пальцев (эритромелалгия). Часто наблюдается гепатомегалия, у 75% пациентов встречается спленомегалия (иногда резко выраженная).
Тромбоз может вызывать симптомы в пораженном участке (например, неврологическая патология при инсульте или транзиторных ишемических атаках, боль в ногах, отек ног или и то и другое при тромбозе сосудов нижних конечностей, односторонняя потеря зрения при тромбозе сосудов сетчатки).
Кровотечения отмечаются у 10% пациентов.
Ускоренный метаболизм может вызывать субфебрильную температуру и приводить к потере веса, что указывает на переход заболевания в фазу истощения. Последняя клинически неотличима от первичного миелофиброза.
Диагностика истинной полицитемии
- Общий анализ крови.
- Тестирование на мутации гена JAK2.
- В некоторых случаях — исследование костного мозга и определение плазменной концентрации эритропоэтина.
- Применение критериев ВОЗ.
Подозрение на ИП часто возникает уже на этапе общего анализа крови, однако оно также должно возникать при наличии соответствующих симптомов, в частности синдрома Бадда-Киари (стоит, однако, отметить, что у некоторых пациентов синдром Бадда-Киари развивается до повышения гематокрита). Нейтрофильный лейкоцитоз и тромбоцитоз являются частыми, но не обязательными проявлениями. Пациенты с изолированным увеличением уровня гемоглобина или эритроцитозом также могут иметь ИП, однако в таких случаях в первую очередь следует исключить вторичный эритроцитоз. ИП также можно заподозрить у некоторых пациентов с нормальным уровнем гемоглобина, но с микроцитозом и признаками дефицита железа. Эта комбинация признаков может возникать при гемопоэзе, протекающем на фоне ограниченных запасов железа, что является отличительной чертой некоторых случаев ИП.
ВОЗ были разработаны новые критерии диагностики. Таким образом, пациенты с подозрением на ИП обычно должны быть протестированы на наличие мутаций гена JAK2.
Исследование образца костного мозга не всегда обязательно.
В тех случаях, когда оно проводится, в костном мозге обычно обращают на себя внимание панмиелоз, большая величина и скученность мегакариоцитов. В некоторых случаях обнаруживаются ретикулиновые волокна. Однако никакие изменения костного мозга не позволяют с абсолютной уверенностью отличить ИП от других патологических состояний (например, врожденной семейной полицитемии), сопровождающихся эритроцитозом.
Концентрация эритропоэтина в плазме у пациентов с ИП обычно низкая или находится у нижней границы нормы. Повышенная концентрация указывает на вторичную природу эритроцитоза.
В некоторых случаях проводится исследование на эндогенное формирование колоний эритроидных клеток в пробирке (предшественники эритроцитов, взятые из периферической крови или костного мозга пациентов с ИП, в отличие от таковых у здоровых людей могут формировать эритроидные клетки в культуре без добавления эритропоэтина).
Определение совокупной массы эритроцитов при помощи эритроцитов, меченных хромом, может помочь отличить истинную и относительную полицитемию, а также отличить полицитемию от миелопролиферативных заболеваний. Однако техника выполнения этого теста сложна. Обычно он не проводится,учитывая его ограниченную доступность и тот факт, что он стандартизирован для использования только на уровне моря.
К неспецифическим отклонениям лабораторных показателей, которые могут отмечаться при ИП, относится повышение концентрации витамина В12 и увеличение В12 — связывающей способности, а также гиперурикемия и гиперурикозурия (присутствуют у >30% пациентов), повышенная экспрессия гена PRV-1 в лейкоцитах, снижение экспрессии гена C-mpl (рецептора тромбопоэтина) в мегакариоцитах и тромбоцитах. Эти тесты не обязательны для установления диагноза.
Диагностика полицитемии обсуждается в подразделе «Повышенное содержание гемоглобина». Для диагностики важно увеличение массы эритроцитов при отсутствии причин для вторичного эритроцитоза и спленомегалия. Число нейтрофилов и тромбоцитов часто увеличено, в костном мозге можно обнаружить патологический кариотип, и in vitro культура костного мозга демонстрирует автономный рост при отсутствии добавления факторов роста.
Прогноз истинной полицитемии
В целом ИП сопряжена с укорочением продолжительности жизни. Медиана выживаемости всех пациентов составляет от 8 до 15 лет, хотя многие живут гораздо дольше. Частая причина смерти — это тромбоз. Следующими по частоте идут осложнения миелофиброза и развитие лейкоза.
Средняя выживаемость после установления диагноза у больных, получающих лечение, превышает 10 лет. Некоторые больные живут более 20 лет; однако цереброваскулярные и коронарные осложнения возникают у 60% больных. Заболевание может перейти в другое миелопролиферативное нарушение; миелофиброз развивается у 15% больных. Острый лейкоз появляется, главным образом, у пациентов, получавших радиоактивный фосфор.
Лечение истинной полицитемии
- Лечение аспирином,
- Возможное кровопускание,
- Возможная миелосупрессивная терапия.
Терапия должна быть подобрана индивидуально с учетом возраста, пола, состояния здоровья, клинических проявлений и результатов гематологических исследований. Пациентов разделяют на группу высокого риска и группу низкого риска. К группе высокого риска относят пациентов >60 лет с анамнезом тромбоза или транзиторных ишемических атак, либо и того,и другого.
Аспирин. Аспирин снижает риск возникновения тромбоза. В связи с этим пациенты, которым проводятся только кровопускания или кровопускания, должны получать аспирин. Более высокие дозы аспирина сопряжены с недопустимо высоким риском кровотечений.
Кровопускание. Кровопускание было основой лечения пациентов в группах как высокого, так и низкого риска, поскольку эксперты считали, что оно снижает вероятность тромбоза. Обоснованность кровопусканий в настоящее время неоднозначна, поскольку новые исследования указывают на то, что уровень гемоглобина может не коррелировать с риском тромбоза. Некоторые клиницисты больше не придерживаются строгих рекомендаций в отношении кровопусканий. Кровопускание по-прежнему остается одной из возможных альтернатив для любого пациента. У незначительной доли пациентов с гиперемией кожи и повышенной вязкостью крови кровопускание может уменьшать выраженность симптомов. Стандартный пороговый уровень гематокрита, выше которого проводится кровопускание, составляет >45% у мужчин и >42% у женщин. Как только величина гематокрита становится ниже пороговой, она проверяется ежемесячно и поддерживается на одном и том же уровне путем дополнительных кровопусканий, которые выполняют по мере необходимости. Если необходимо, внутрисосудистый объем восполняется кристаллоидными или коллоидными растворами.
Миелосупрессивная терапия показана пациентам из группы высокого риска.
Радиоактивный фосфор (32Р) долгое время использовался для лечения ИП. Эффективность лечения составляет от 80 до 90%. Радиоактивный фосфор хорошо переносится и требует меньшей частоты посещений клиники после достижения контроля над заболеванием. Однако применение радиоактивного фосфора сопряжено с увеличением риска развития острого лейкоза. Лейкоз, возникающий после такой терапии, часто резистентен к индукционной терапии и всегда неизлечим. Таким образом, применение радиоактивного фосфора требует тщательного отбора пациентов (например, препарат следует назначать только тем пациентам, чья ожидаемая продолжительность жизни в связи с сопутствующей патологией не превышает 5 лет). Назначать его стоит только в редких случаях. Многие врачи не используют его вовсе.
Гидроксимочевина подавляет фермент рибонуклеозиддифосфат-редуктазу. Она также используется для подавления активности костного мозга. Однозначных данных о способности гидроксимочевины провоцировать лейкоз нет. Однако вероятность трансформации в лейкоз существует, хотя она и мала. Пациентам еженедельно выполняют анализ крови. После достижения равновесного состояния интервалы между анализами крови увеличивают до 2 недель, а затем до 4 недель. Если уровень лейкоцитов падает <4000/мкл или уровень тромбоцитов падает <100 000/мкл, лечение приостанавливают, а когда упомянутые показатели приходят в норму, возобновляют в дозе на 50% меньше исходной. Дозу гидроксимочевины рационально титровать до достижения практически нормальной величины гематокрита, однако данные в пользу такого титрования отсутствуют. Нормализация уровня лейкоцитов, вероятно, более важна, но как и в предыдущем случае, эта гипотеза не была подтверждена проспективными исследованиями. Подтверждения тому, что нормализация уровня тромбоцитов необходима, нет, и некоторые врачи не увеличивают дозу гидроксимочевины до тех пор, пока число тромбоцитов остается <1,5 млн/мкл. Острая токсичность — нередкое явление. В некоторых случаях у пациентов возникает сыпь, лихорадка, изменения внешнего вида ногтей, кожные язвы.
Интерферон альфа-2b применяется в тех случаях, когда с помощью гидроксимочевины не удается поддерживать нужный уровень форменных элементов крови или при неэффективности последней. Стоит отметить, что пегилированный интерферон альфа-2Ь обычно хорошо переносится. Этот препарат воздействует на заболевание на молекулярном уровне и обладает относительно низкой токсичностью.
Алкилирующие препараты могут провоцировать развитие лейкоза, поэтому их стоит избегать.
В настоящее время в фазе клинической разработки находится несколько ингибиторов каскада JAK2. В основном они исследуются у больных с поздними стадиями миелофиброза.
Лечение осложнений. Гиперурикемию корректируют с помощью аллопуринола, если повышение концентрации мочевой кислоты сопровождается симптомами или если пациенты одновременно получают миелосупрессивную терапию. Зуд можно пытаться контролировать с помощью антигистаминных препаратов, однако иногда этого трудно добиться. Миелосупрессия часто является наиболее эффективным методом. Примером потенциально эффективной терапии может быть холестирамин, ципрогептадин, циметедин или пароксетин.
Кровопускание быстро купирует симптомы повышенной вязкости. Удаляют 400-500 мл крови — и венесекцию повторяют каждые 5—7 сут до снижения гематокрита ниже на 45%, удаляя при каждой процедуре 400—500 мл крови (меньше, если больной пожилой). Менее частые, но регулярные кровопускания, поддерживают этот уровень, пока гемоглобин не станет меньше из-за дефицита железа. Лежащую в основе заболевания миелопролиферацию подавляют гидроксикарбамидом или интерфероном. Лечение радиоактивным фосфором (5 мКи 32Р внутривенно) оставляют для пожилых пациентов, так как оно в 6—10 раз повышает риск трансформации в острый лейкоз. Лечение пролиферации костного мозга может уменьшить риск окклюзии сосудов, контролировать размеры селезёнки и уменьшить трансформацию в миелофиброз. Аспирин снижает риск тромбозов.
www.sweli.ru
Оставить комментарий